
One of the scariest requests you may hear from your R&D manager is “please join us for an FMEA meeting today after lunch”. Your first reaction would probably be something like “I didn’t do it” much like when you’re called into the high school principal’s office knowing you’re in trouble even though if you didn’t do anything. For those of us who are unfamiliar with the term, FMEA stands for Failure Mode and Effect Analysis. The English translation of these 5 words is what could go wrong in our product or production process such that its performance (and very rarely also yield in the case of process) does not meet our requirements. It is, so to speak, a different way of asking what are our product risks? Note that this is different to the project risks that we covered in a separate post series. Project risks are what may happen for the project to fail while FMEA covers the failure, excluding market risks, of the product itself.
So, if the R&D manager suddenly decides to hold an FMEA meeting it usually means that something went seriously wrong with the product itself and now we have to find which erroneous situation, or heaven forbid, what hazard slipped by in our design or production process and materialized in the client’s place.
Who, When and Why FMEA?
An FMEA is an integral part of product design which is done continuously throughout the development process. Generally, there are two kinds of FMEAs that relate to different stages in the product life cycle: Design stage risks (DFMEA) and Process stage risks (PFMEA). In this post I will refer to FMEA in general unless it specifically concerns only one these two FMEAs.
The why, should be obvious and straight forward: what could go wrong in our product, but there is a bit more to it. The outcome of an FMEA process is a thorough report of what can go wrong in our product and how we handle it within our product operation, manufacturing, service or any other acceptable way. It serves a dual purpose: internally within our company to verify that we have covered all the erroneous situations that may occur and more importantly externally to present it to the client, regulatory authority or any other party that requires this process for their product approval and compliance.
And finally, the who. You guessed it ![]() – the System Engineer has to be in charge of the process, with a lot (and I mean a lot) of help from all other disciplines technical and non-technical. In the cases where there are different System Engineering teams for R&D and Production\Sustain, both should be highly involved in the FMEA and according to the specific product stage in the life-cycle they should decide who among them has to lead the process.
– the System Engineer has to be in charge of the process, with a lot (and I mean a lot) of help from all other disciplines technical and non-technical. In the cases where there are different System Engineering teams for R&D and Production\Sustain, both should be highly involved in the FMEA and according to the specific product stage in the life-cycle they should decide who among them has to lead the process.
The FMEA Process
FMEA process may go bottom-up or top-down, but the best and most thorough method is to have them both done in two different paths.
Bottom-up method takes each and every component in the system and maps what could go wrong in this component. For example, if we have a temperature sensor it may get burnt, it may show deviated temperatures, it may cause an electric short in the electric system and so on. After mapping each and every failure mode {aha!!! you see? There is merit in that obscure acronym ![]() }, we follow the path upwards to see how that failure affects the system’s performance.
}, we follow the path upwards to see how that failure affects the system’s performance.
Top-down will be taking our System Requirements one by one and simulating non-compliance with that component. Here we will be breaking each requirement down one-by-one to its sub-components in our implementation to identify which components affect which System Requirement upwards to cause that non-compliance. For example, if our product is a furnace that has to reach, but not exceed, the temperature 275oC then non-compliance with that requirement may be caused by a faulty temperature sensor.
For each such scenario we also assess the probability of it to occur. Taking the same example, our temperature sensor gets burnt in 1:100,000 units during the period of 15,000 hours of operation which is the expected lifetime of the sensor however, the same sensor may deviate above the accepted limit after 10,000 hours of operation in 1:5,000 units.
Immediately after doing one or both methods we go over each scenario and assess what the handling of this erroneous situation is. Is the error detected? Is it automatic? Does it require an involvement of the user? Is it recoverable?
So eventually we would have a list of things that may go wrong, what their probabilities are of occurring and how we detect them.
Error Lists
Over the years I discovered that the most efficient FMEA processes were when there was a systematic error list which covers everything that may go wrong in the system level and if it is caught by some SW error, and if so, which error it raises.
I must admit that creating and maintaining such a list is a tedious process but it is worth it and for all the right reasons: the FMEA process goes much smoother, post-integration and first customers introduction have quicker responses when problems arise, service manuals eventually become more comprehensive, the SW infrastructure and error handling mechanism are more profound and many more good reasons.
Of course, if we take the error lists one step forward, we could also have the error handling for each SW error.
Product Risks
We cannot talk about FMEA without really mentioning what are product risks. There are many risks around product usage and operation but all can be summarized into 4 main categories:

Hazard to User
Situations, where the user may end up in the emergency room or in extreme circumstances in the cemetery, are considered hazards to the user. The term user refers to any person, who in reasonable use, uses or is being used upon with our system. It means that the FMEA has to include the case of a service engineer who opens up the covers of a laser machine and being exposed to high power laser even though the machine is meant to be operated in closed covers by non-service users. However, the case of a user that takes the electricity jack and disassembles it and while doing so gets electrocuted, is not a case of reasonable use (it may even be considered as abusive usage). I will share a personal experience here: I visited a client who had 3 of our machines in a row and for my comfort I leaned on 2 machines. Immediately I took in a surge of 230V AC for a split second that left me shaking for 30 seconds before I was able utter a sound. After recovering I started to look around and check how on earth our machine could get high voltage leakage through the plastic cases. Apparently, the main electrician of that facility had decided that when you connect 3 machines in a row you do not require grounding and as such all 3 machines were electrically floating although all were kept with their original standard 3 pin electricity jack. Obviously if you accidentally touched 2 machines at once you get electrocuted because the screws connecting the plastics were metallic. Now this specific case was NOT covered when we mapped our hazards since we could not imagine someone creative enough to the point of not connecting the grounding of the machine. That is considered as an abusive usage.
We have to remember though that even if it is not our intended use for the machine, if there is a use case in which a substantial number of users operate the machine it must be covered in the FMEA.
Hazard to the Environment
Hazard to the environment is the risk of having an effect on the environment but does not inflict direct harm to the user or to our system. For example, if our system constantly emits gas that is harmful for the ozone layer.
BTW, hazards to users and the environment are covered by ISO and other standards. If the compliance with such standards is part of your product requirements, then you could consider them as “Non-compliance with Requirements”. Nevertheless, I would treat both Hazards to the User and to the Environment with extra care regardless of which standards are in our “to-comply” list.
Remember we are not lawyers. If there is a substantial risk of finger amputation by our machine, even if it is not an “official” requirement and there is no relevant standard that addresses this issue, we wouldn’t want anyone to be rushed into the hospital because of our machine malfunctioning.
Hazards to the System
Hazard to the system is any situation after which the system will be non-operational. The best example I could give for such a hazard is lighting protection in the entrance power-supply of a machine, in order to protect the delicate electronics behind it.
Hazards to the system usually arise when doing the bottom-up FMEA approach.
Non-compliance with Requirements
The risk of non-compliance with the product requirement is simple – the system does not do what it is supposed to do. This is of course the most general family of product risks since it may include any requirement for any product. But, to remind you, in the top-down FMEA process we cover this exact family of risks in full, so having an FMEA process done in an orderly manner will surely get us to the right place.
Types of Detection and Error Handling
There are 3 types of detection of failures:
Automatic – the system automatically detects the failure such that either the SW or the HW has an indication of a failure happening.
Manual – the user gets an indication of the failure but it cannot be sensed by the system. For example, if a screen presentation gets stuck but the all the display drivers are responding.
Undetected – the system continues running as if everything is OK, but its performance is not up to requirements. Undetected will always be followed by unhandled as there is no indication of the problem. This is the most dangerous situation and has to be minimized to zero in our systems.
Accordingly there are 3 types of failure handling:
Automatic – error recovery happens automatically by the system. That could be done on automatic detection, or manual detection where the user activates the automatic handling.
Manual – manual handling means that a person has to actively go through a series of actions to fix the problem (pressing a button is excluded to allow the automatic handling on a manual detection). For example, an optical system’s alignment is off and needs manual fixing of lens locations.
Unhandled – the error is either registered to a log (in the cases of automatic or manual detection) or nothing is done to recover. Acceptable or not, that is for the team to decide according to the severity of the failure.
When addressing the fault detection and handling we should also address the latency of the flow of events. If processing of the data regarding our furnace arrives 5 minutes after the furnace has overheated, we’ve already burnt our pizza and the same goes for any hazard to the user. In real-time applications the latency cannot be overlooked and should be part of the FMEA considerations.
Eventually we have to quantify the severity of each product risk in the FMEA, very much like in the project risk assessment in which we define a metrics. The method is similar to project risk with a similar table but here we have to take into account 3 variables: risk severity, probability of occurrence and detectability. Again, taking the furnace example, we could have a simple metrics table like the following table. Note that the cause was also referenced for readability ease, and in order to have some kind of system engineering best practices I added an additional sensor for redundancy:
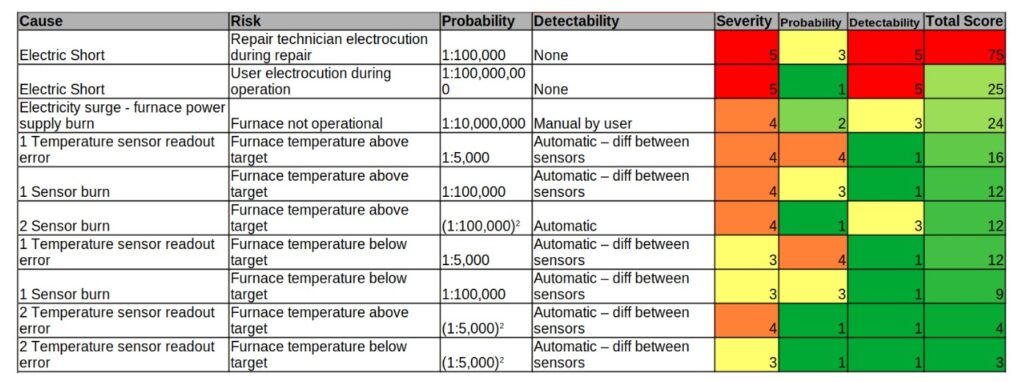
There are many versions to this kind of table, sometimes with columns such as prevention, latency and backup strategies and many more – this is really up to the System Engineer how much data to squeeze into this table.
Medical Industry
This post would not be complete without slipping in a word or two about Medical devices.
In the medical world FMEA and risk management are a big deal as the outcome is submitted to the regulatory authorities as part of the official submission. Obviously the FMEA process in this field has extra importance. No one can allow having a stent failing any of its requirements before, during or after a catheterization procedure and we may deduct the same for any other medical device, whatever its purpose is. These are the main reasons why FMEA and risk management processes are so important. In addition, take note that unlike other disciplines in medical devices, the personnel that are involved in these processes would be the extended team which includes almost everyone in the team from R&D and product through clinical affairs and medical experts and up to regulatory affairs and legal department.
One other note before wrapping up: no matter how good your FMEAs will be you will get unexpected failures in the field, at least in the beginning of the product life. They are bound to happen due to the complexity of our systems and the complexity of the environments we operate in. Don’t be discouraged to the point of skipping or disrespecting the FMEA process. Look at them from the brighter side or things: it could have been much worse if you hadn’t performed the FMEA properly.
Of course, there is much more to the FMEA. PFMEA is one of many steps to reach six sigma and can be a main base activity for yield improvements. A proper DFMEA, when started early on after the concept stage in the development process, may save the costs of error handling mechanism addition in the late stages of the development. It is not just another task of the System Engineer as its results may impact the whole design, manufacturing, assembly, installation and even distribution processes of the product. When you think about it, it is very much like simulation games where we can play with “what happens if” but of an engineering purpose. Try to enjoy it; if you put the little details aside for a moment, it is a fun process like a maze.